This web page was produced as an assignment for Genetics 564, a capstone course at UW- Madison.
What is Hemophagocytic Lymphohistiocytosis?
Familial Hemophagocytic Lymphohistiocytosis (fHLH) is a genetic condition characterized by excessive activation and multiplication of white blood cells. This causes macrophages and T-cells, the specific types of white blood cells affected, to damage target organs as well as release cytokines, which signal inflammatory responses [7].
Hemo-Relating to 'Blood' Lympho-Specifies this is a condition affecting the lymphatic system, part of the immune system |
PhagocyticRefers to phagocytes, a type of white blood cell that engulfs foreign microorganisms and other materials. Together, 'Hemophagocytic' means that these phagocytes are damaging blood producing bone marrow cells [5]. HistiocytosisReferring to a condition that causes an abnormal amount of histiocytes, cells derived from WBCs [1,4]. |
'Familial' HLH suggests that this condition can be inherited. More specifically, 'familial' HLH differs from 'genetic' HLH in that 'familial' HLH is the affect of a biallelic mutation variants [7]. There are cases of HLH that are not inherited and generally are a reaction to some trigger [4]. This website focuses on the genetic condition.
What is the Inheritance pattern? fHLH often appears as an autosomal recessive disorder[7], meaning a mutated allele from both parents is required to have the condition. |
What are the symptoms?
Diagnosis of fHLH requires the identification of the two mutated alleles associated with the condition. This would be completed with either a multigene panel, which can identify multiple gene causes, or genomic sequencing methods. This must pair with clinical diagnosis [7*]. Three criteria for considering fHLH diagnosis:
|
Fever
Prolonged and high fevers are indicative of fHLH[2]
|
Hepatosplenomegaly
Enlarged spleen and liver are symptoms of fHLH patients [2]
|
Cytopenias
Lower than average neutrophils, hemoglobin, and platelets are observed in fHLH patients [7]
|
Additional symptoms include: Rash, Swollen lymph nodes, neurological symptoms, and, less commonly, acute liver failure and Central Nervous System involvement without presence of other symptoms [7].
Incidence + Prevalence
This condition is rare, affecting about 1 in 50,000 individuals around the world [4]. Diagnosis generally occurs within the first few months of an individuals life, but there have been situations of diagnosis in childhood or adulthood. Life expectancy and course of the disease are unknown in FHLH diagnosed adults [7].
What is PRF1?
PRF1 is one of the genes that is associated with fHLH. This gene is located on chromosome 10 [5]. 30-40% of fHLH is the result of a biallelic inheritance of mutated PRF1 [7].
What is perforin?
PRF1 encodes for the protein perforin, which is associated with the cell destruction process of the immune system, known as cytolysis [4]. Cytolysis involves the release of lysosomes from natural killer (NK) and T-cells at a targeted cite. In cytolysis, perforin's job is to create a pore on the targeted cell for the granzymes and other contents to induce apoptosis (cell death) [2,7]
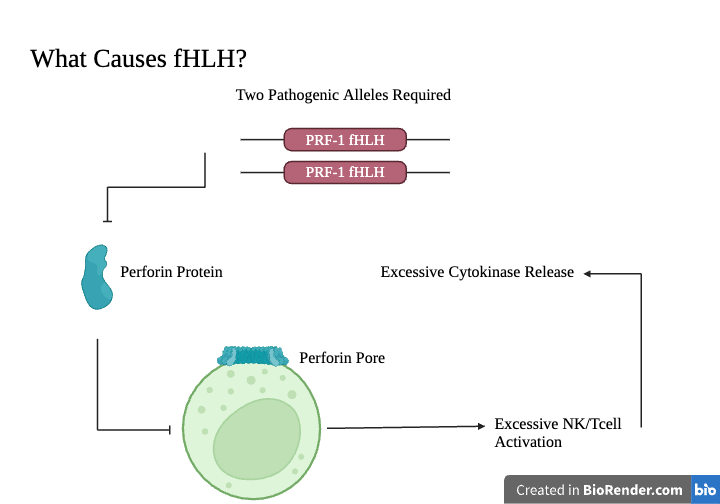
How does Perforin Mutation cause symptoms?
|
The loss of function mutation in the two PRF1 alleles results in inhibited perforin production. This leads to an inability for granzymes to enter the targeted cell and initiate apoptosis. Th1 cells initiate an immune response, activating macrophages and more NK and T-cells. Excessive cytokines are then released, causing inflammatory response [3]. |
CHeck out the Histiocyte society for further information and resources
References
